Elemental impurities in drug products may arise from several sources (e.g. residual catalysts employed during the synthetic process of the drug substance, interaction of the final product with manufacturing equipment or the container closure system, etc.). Since elemental impurities do not provide any therapeutic benefit to the patient, their levels in the drug product should be controlled within acceptable limits. To this end, ICH Q3D presents a process to assess and control elemental impurities in the drug product using the principles of risk management.
The risk assessment process can be described in three steps:
- Identify known and potential sources of elemental impurities that may find their way into the drug product.
- Evaluate the presence of a particular elemental impurity in the drug product by determining the observed or predicted level of the impurity and comparing with the established PDE.
- Summarize and document the risk assessment. Identify if controls built into the process are sufficient or identify additional controls to be considered to limit elemental impurities in the drug product.
The first step of the risk assessment process comprises of the identification of known and potential sources of elemental impurities that may find their way into the final product. The figure below illustrates potential sources that will be considered during the evaluation.
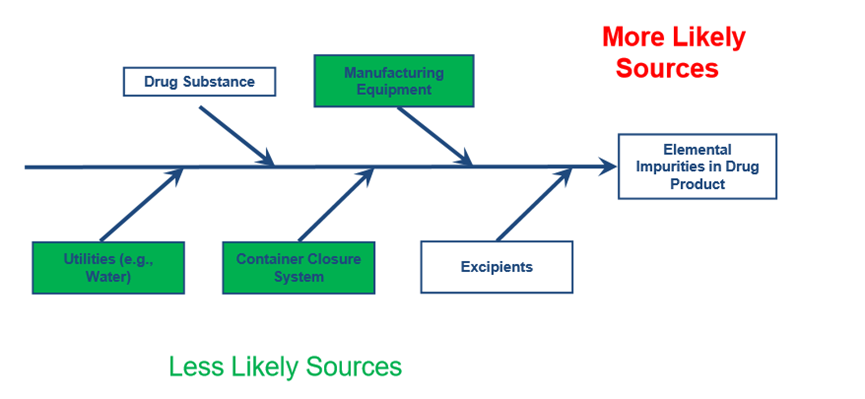
A Failure mode and effect analysis (FMEA) is conducted in order to identify and assess the risk associated with each potential source of elemental impurities. FMEA is a highly structured, systematic technique for failure analysis. It involves the review of all components depicted in the Figure above in order to identify failure modes and their effects. For each component, their failure modes and their resulting effects are recorded in a FMEA sheet. Each failure mode is associated with a material, process or parameter that could serve as a source of elemental impurities. The failure effects correspond to the metals that could emanate from each source. The degree of severity of the effects, their respective probabilities of occurrence, and their detectability are assessed by assigning appropriate numerical values. A Risk ranking and filtering approach is subsequently adopted in order to compare and rank risks. The hybrid FMEA and Risk ranking and filtering methodology aims at facilitating the investigation and the subsequent establishment of additional measures / controls in case the elemental impurity levels exceed the control threshold ( 30% of the PDE). The identification process is based on the review of the API, excipient and drug product manufacturing process to identify known and potential sources of Elemental Impurities. The elements that have to be considered are Class 1 and 2A metals, 2B in case they have been intentionally added, and class 3, depending on the route of administration. In all cases, all intentionally added elements have to be assessed.
The next step is the evaluation of the presence of particular elemental impurities in the drug product. The latter is based on the collection of predicted and/or observed levels of elemental impurities and the subsequent comparison of data with the established Permitted Daily Exposure. The data to support the evaluation may emanate from published literature, data generated from similar processes, supplier information data (e.g Certificates of Analysis), testing of the components of the drug product or the final product itself.
The final step comprises of the documentation and summary of the risk assessment and the identification of additional control requirements if needed.
The proper implementation of the guideline requires a combination of a scientifically sound risk-assessment and testing of the final product and/or components. Based on the amount and quality of available data one could minimize the required testing. However, the level and variability of an elemental impurity should be well established. The latter means that in the absence of other justification, data from testing 3 representative production scale lots or 6 representative pilot scale lots of the component(s) or drug product would be required.



